![]()
Displasia Tanatofora OMIM 187600 - OMIM 187601
Displasia Tanatofora OMIM 187600 OMIM 187601
E la forma più frequente di displasia scheletrica letale neonatale con frequenza doppia nei maschi. Se ne distinguono due forme con caratteristiche ecografiche sostanzialmente diverse.
Genetica: è legata a mutazione del gene che codifica per il recettore del fattore di crescita dei fibroblasti FGFR3; si ha anche una maggiore frequenza con l'età paterna avanzata. I geni FGFR comprendono una famiglia di polipeptidi coinvolti nella crescita e nella differenziazione di varie cellule di origine mesenchimale e neuroectodermica.
Displasia Tanatofora tipo I OMIM 187600
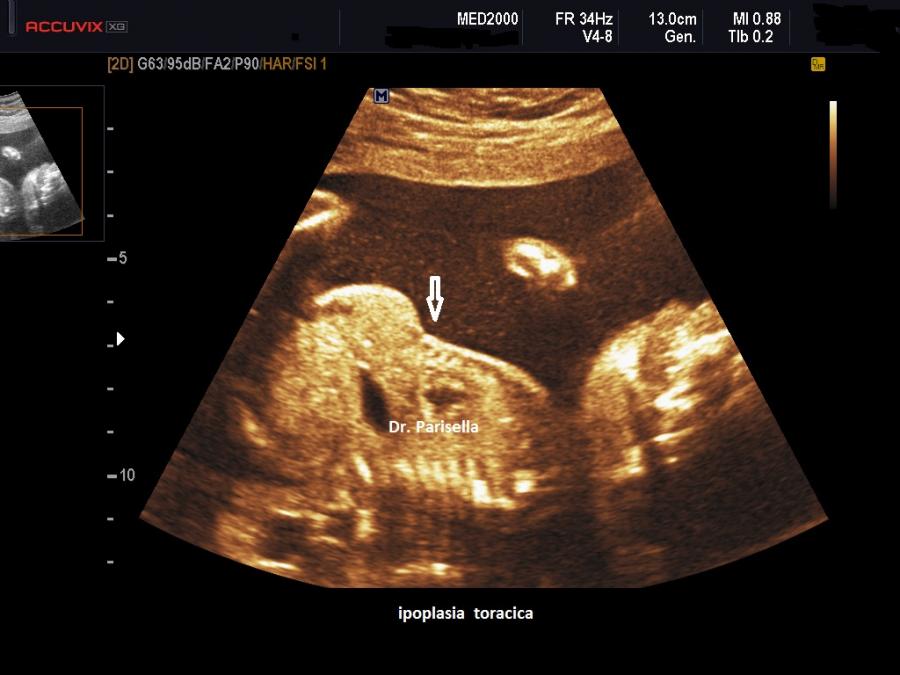
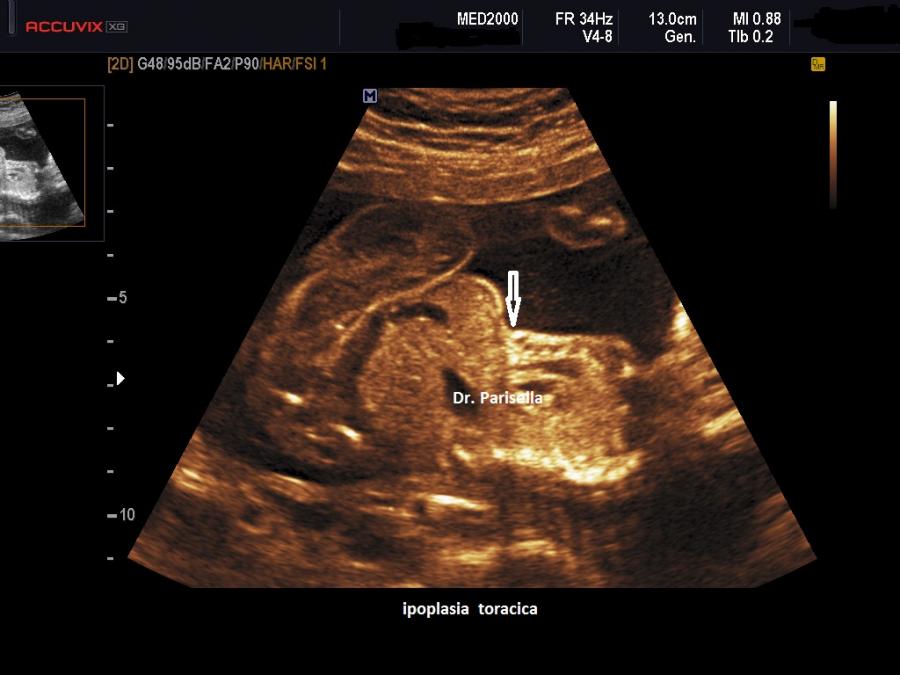
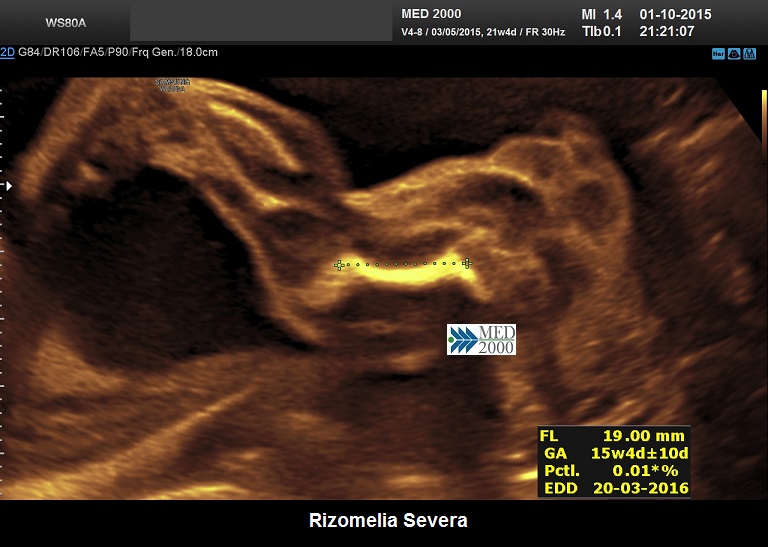
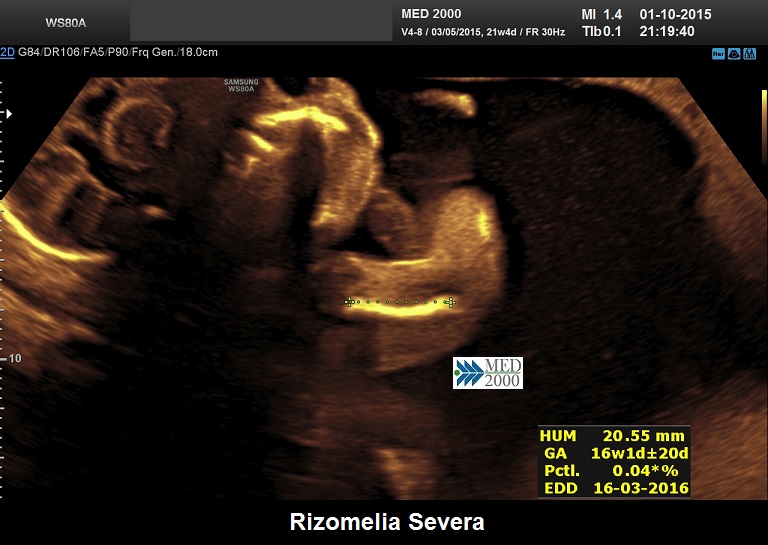
La Displasia Tanatofora tipo 1 è trasmessa con modalità autosomica dominante ed è caratterizzata principalmente da RIZOMELIA SEVERA, FEMORI CORTI (A CORNETTA DI TELEFONO), IPOPLASIA TORACICA SEVERA.
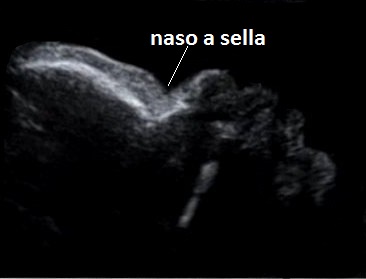
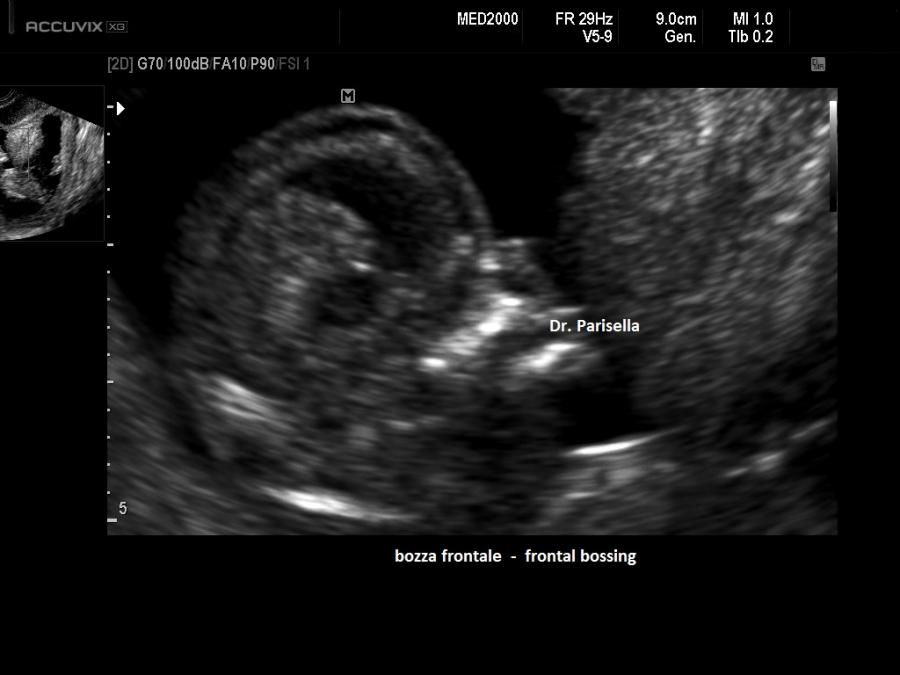
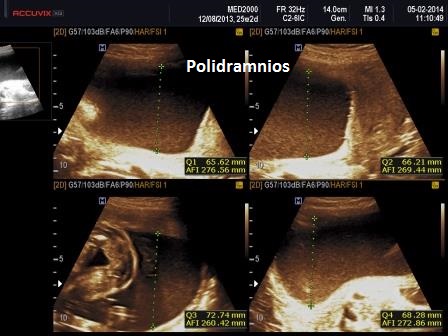
Dal punto di vista ecografico il tratto rizomelico è estremamente ipoplasico, ricurvo e con le metafisi slargate (Femore a Cornetta di Telefono) - (il riconoscimento del Femore a Cornetta di Telefono rappresenta un segno importantissimo e sufficiente per porre con certezza la diagnosi di Displasia Tanatofora tipo I); vi sono coste corte con ipoplasia toracica e si osserva il classico gradino al passaggio tra torace ipoplasico e addome apparentemente prevalente; vi è in genere macrocrania con bozze frontali prominenti (frontal bossing) e naso a sella. Si associa polidramnios severo.
DIAGNOSI DIFFERENZIALE:
Prognosi
letale nel 100% dei casi
Rischio di ricorrenza
Essendo dovuta a mutazioni de novo il rischio di ricorrenza è estremamente basso
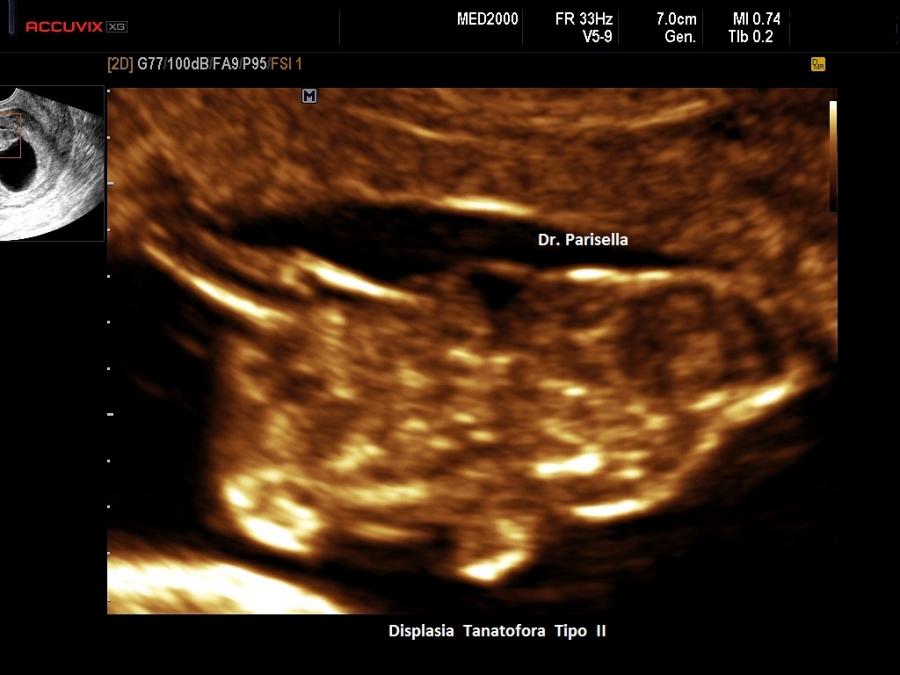
Displasia Tanatofora tipo II OMIM 187601
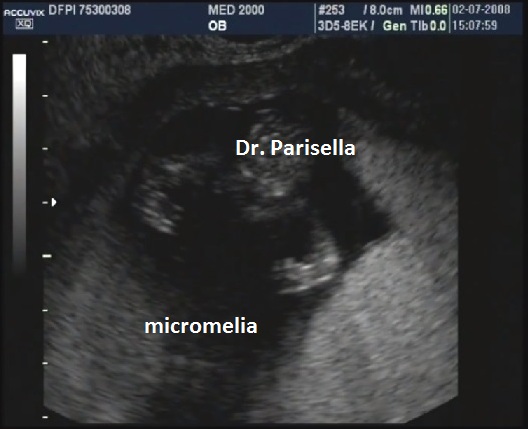
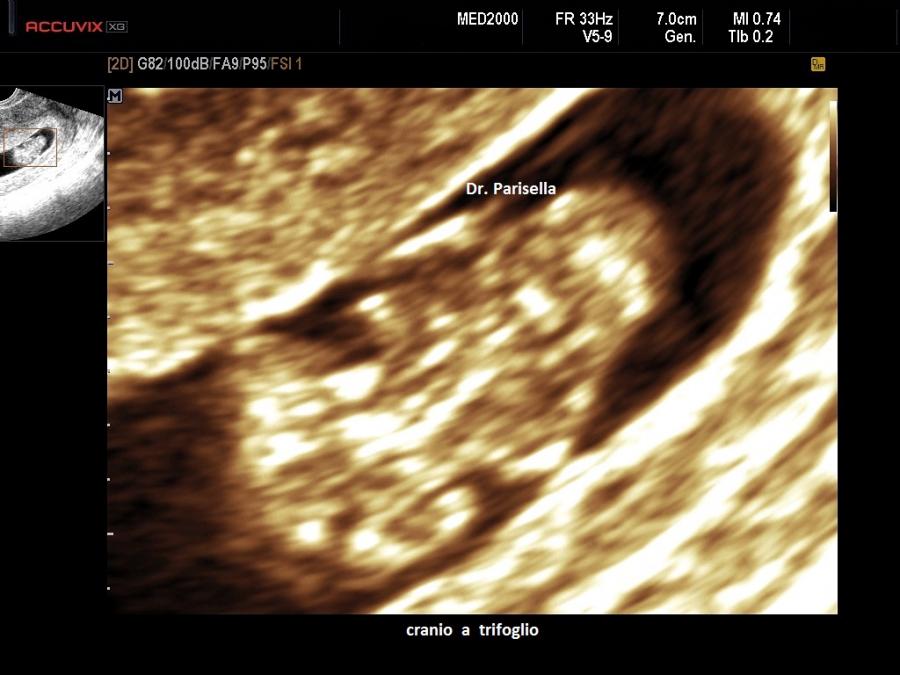
La Displasia Tanatofora tipo II è trasmessa con modalità autosomica dominante ed è caratterizzata da MICROMELIA SEVERA, IPOPLASIA TORACICA SEVERA e CRANIO A TRIFOGLIO.
Dal punto di vista ecografico la presenza contemporanea di MICROMELIA e CRANIO
A TRIFOGLIO rappresentano una associazione di notevole rilevanza diagnostica per questa
patologia.
Il cranio a trifoglio è evidenziabile in scansione coronale ed è dovuto alla
sinostosi delle suture lambdoidea, coronale e sagittale e conseguente
prominenza delle regioni temporali. Caratteristicamente in questi casi si ha molta difficoltà a misurare il Diametro Biparietale.
Le ossa lunghe appaiono meno ipoplasiche e ricurve che nel Tipo I.
Il Polidramnios è ad insorgenza tardiva.
DIAGNOSI DIFFERENZIALE: il cranio a trifoglio può essere presente nella
Sindrome di Pfeiffer (craniosinostosi, ipertelorismo, sindattilia), nella
Sindrome di Crouzon (arti normali, craniosinostosi, ipoplasia mascellare),
nella Sindrome di Apert, nella Sindrome di Carpenter, nella Sindrome di
Kleeblatshadel.
Prognosi
letale nel 100% dei casi
Rischio di ricorrenza
Essendo dovuta a mutazioni de novo il rischio di ricorrenza è estremamente basso.
Baker, K. M., Olson, D. S., Harding, C. O., Pauli,
R. M. Long-term survival in typical thanatophoric dysplasia type 1. Am. J. Med.
Genet. 70: 427-436, 1997.
Camera, G., Dodero, D., De Pascale, S. Prenatal diagnosis of thanatophoric
dysplasia at 24 weeks. Am. J. Med. Genet. 18: 39-43, 1984.
Corsello, G., Maresi, E., Rossi, C., Giuffre, L., Cittadini, E. Thanatophoric
dysplasia in monozygotic twins discordant for cloverleaf skull: prenatal
diagnosis, clinical and pathological findings. Am. J. Med. Genet. 42: 122-126,
1992.
Elejalde, B. R., Mercedes de Elejalde, M. Thanatophoric dysplasia: fetal
manifestations and prenatal diagnosis. Am. J. Med. Genet. 22: 669-683,
1985.
Hall, C. M. International nosology and classification of constitutional
disorders of bone (2001). Am. J. Med. Genet. 113: 65-77, 2002.
Horton, W. A., Hood, O. J., Machado, M. A., Ahmed, S., Griffey, E. S. Abnormal
ossification in thanatophoric dysplasia. Bone 9: 53-61, 1988.
Horton, W. A., Harris, D. J., Collins, D. L. Discordance for the
Kleeblattschaedel anomaly in monozygotic twins with thanatophoric dysplasia.
Am. J. Med. Genet. 15: 97-101, 1983.
Norman, A. M., Rimmer, S., Landy, S., Donnai, D. Thanatophoric dysplasia of the
straight-bone type (type 2). Clin. Dysmorph. 1: 115-120, 1992.
Sawai, H., Komori, S., Ida, A., Henmi, T., Bessho, T., Koyama, K. Prenatal
diagnosis of thanatophoric dysplasia by mutational analysis of the fibroblast
growth factor receptor 3 gene and a proposed correction of previously published
PCR results. Prenatal Diag. 19: 21-24, 1999.
Schild, R. L., Hunt, G. H., Moore, J., Davies, H., Horwell, D. H. Antenatal
sonographic diagnosis of thanatophoric dysplasia: a report of three cases and a
review of the literature with special emphasis on the differential diagnosis.
Ultrasound Obstet. Gynec. 8: 62-67, 1996
Tavormina, P. L., Shiang, R., Thompson, L. M., Zhu, Y.-Z., Wilkin, D. J.,
Lachman, R. S., Wilcox, W. R., Rimoin, D. L., Cohn, D. H., Wasmuth, J. J.
Thanatophoric dysplasia (types I and II) caused by distinct mutations in
fibroblast growth factor receptor 3. Nature Genet. 9: 321-328, 1995.
 0_n2kt0hr2.jpg)
 1_j02w0dnx.jpg)
 00_lw1yylb2.JPG)
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...