![]()
Sindrome di Patau o Trisomia 13
Sindrome di Patau o Trisomia 13
Redatto da: P.Parisella
La trisomia 13 è un'anomalia cromosomica causata dalla presenza di un cromosoma 13 in sovrannumero. L'incidenza è di circa 1/8000-1/15000 nati e nei feti affetti la morte endouterina si verifica in oltre il 95% dei casi.
Il meccanismo che è più frequentemente causa della trisomia 13 è la non disgiunzione dei cromosomi durante il processo di divisione cellulare all'interno del gamete di uno dei genitori; l'ovulo o lo spermatozoo conservano quindi entrambe le copie del cromosoma 13 all'interno della cellula; quando avviene la fecondazione, con l'apporto paterno o materno di un'altro cromosoma 13, si formano cellule trisomiche cioè con tre copie del cromosoma 13.
In una percentuale minore la causa può essere una traslocazione, prima o al momento del concepimento, di materiale extra del cromosoma 13 ad un'altro cromosoma; quindi le persone affette hanno due copie del cromosoma 13 più materiale extra dal cromosoma 13 attaccato ad un'altro cromosoma. La traslocazione porta ad una trisomia 13 parziale per cui i segni clinici della malattia possono essere più sfumati.
Possono esservi anche forme in cui la traslocazione può essere ereditata per la presenza di una traslocazione bilanciata in uno dei genitori; nella traslocazione bilanciata non vi è materiale extra dal cromosoma 13 ma solo un riarrangiamento di materiale genetico tra il cromosoma 13 ed un'altro cromosoma; i portatori di questo tipo di traslocazione non hanno segni della malattia ma hanno un aumento del rischio di concepire un figlio affetto.
In una piccola percentuale dei casi si hanno forme di mosaicismo: in questi casi l'espressione fenotipica può essere assente o più o meno marcata in base al numero di cellule trisomiche presenti nei tessuti.
DIAGNOSI
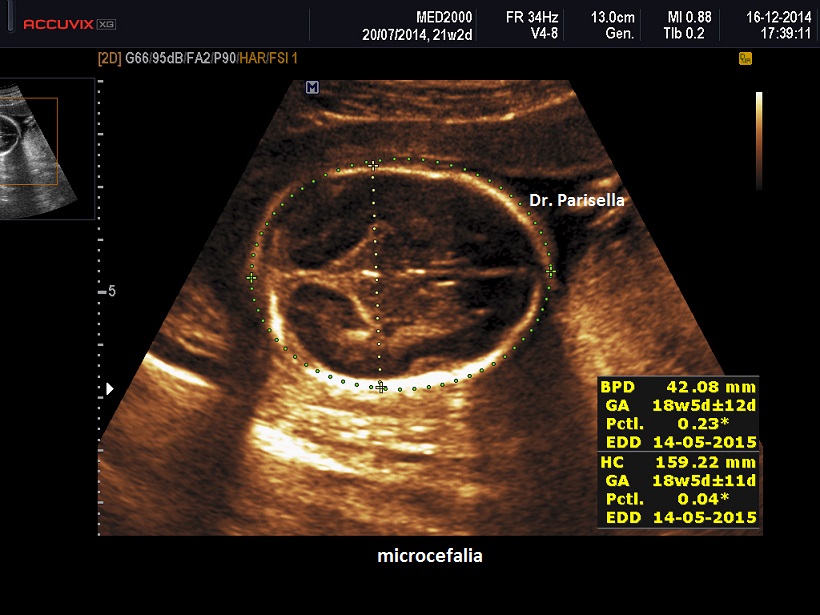
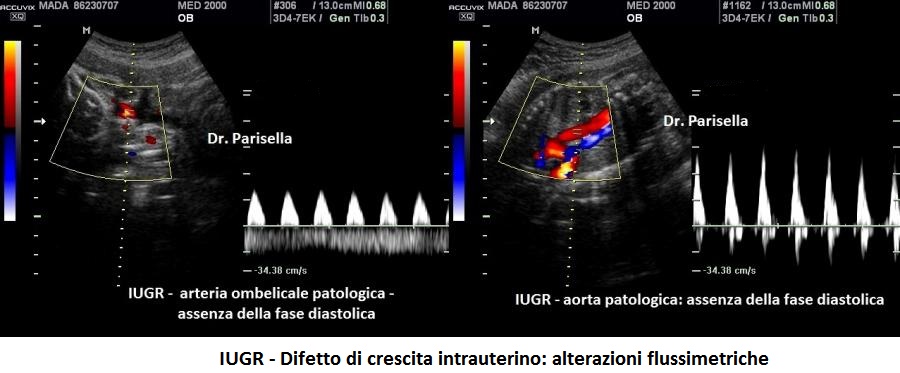
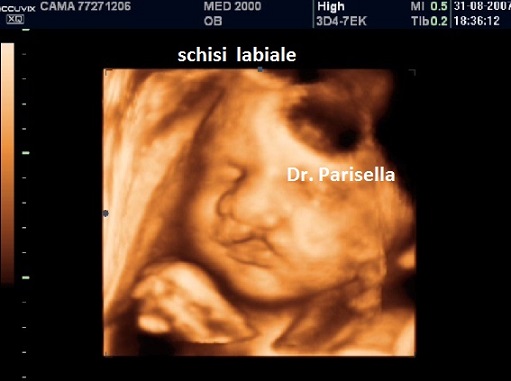
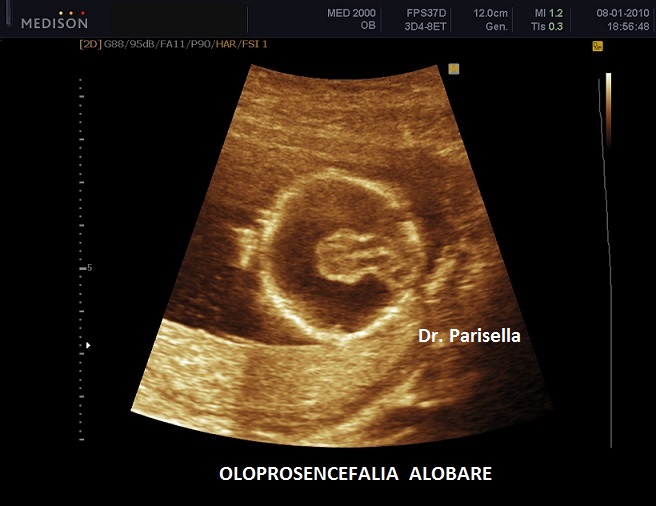
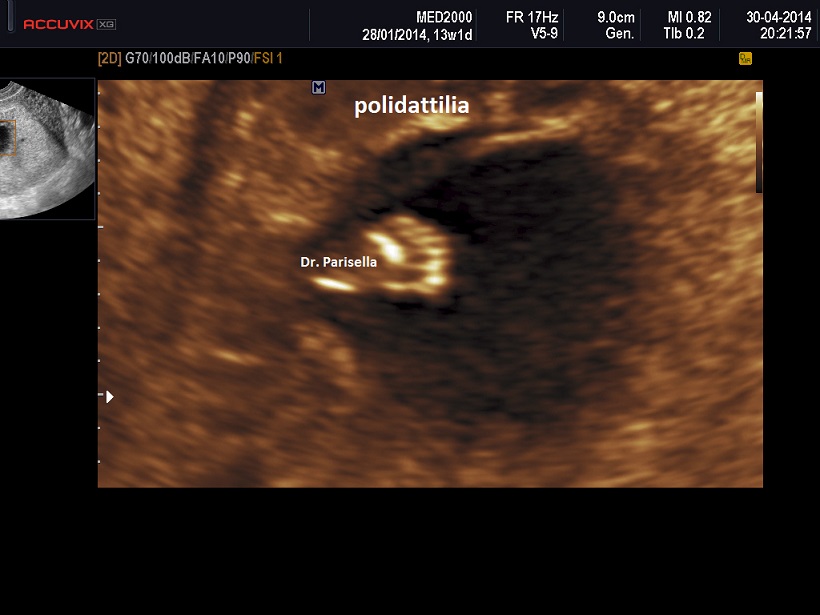
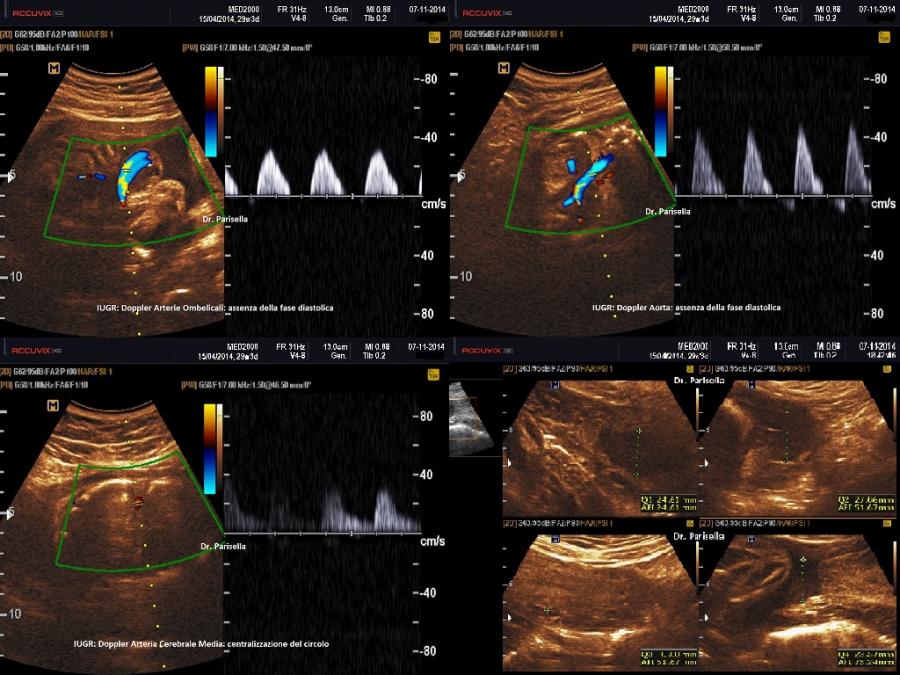
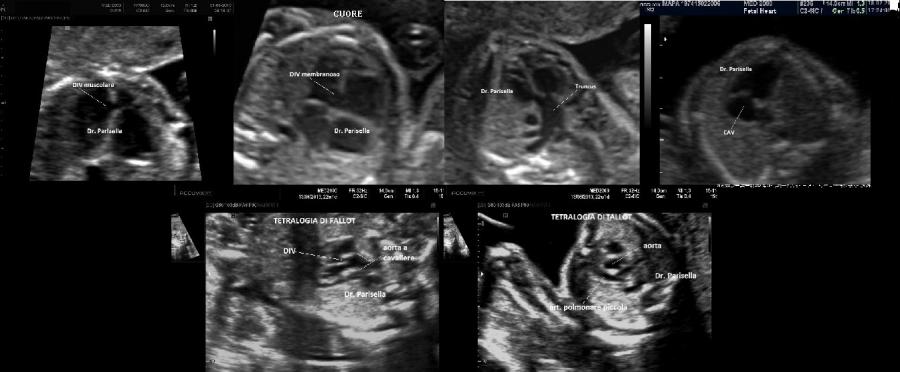
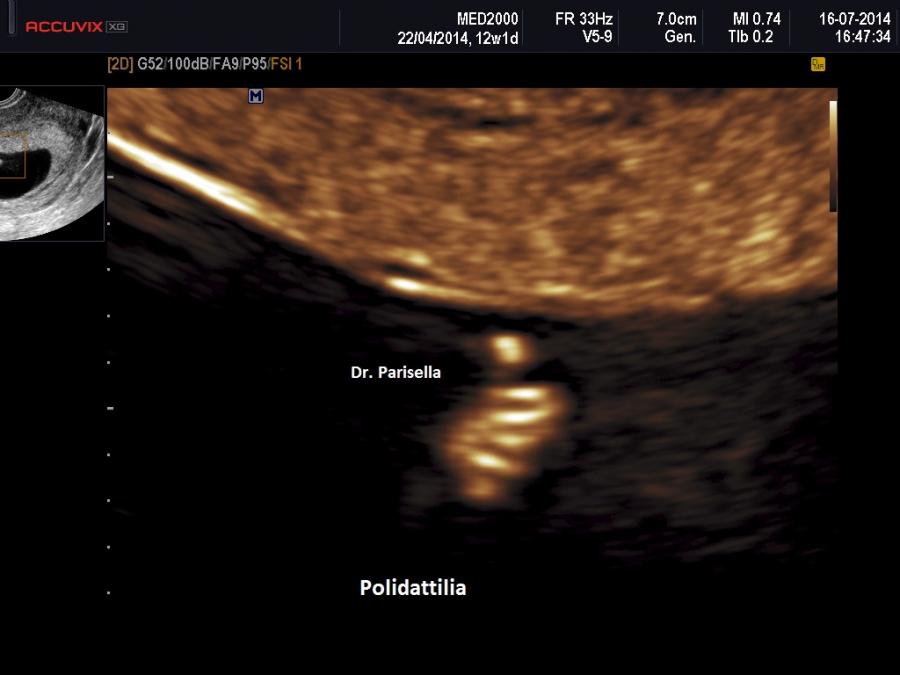
E' caratterizzata principalmente da IUGR precoce, oloprosencefalia, polidattilia postassiale, microcefalia, anomalie facciali (anoftalmia o microftalmia, ipotelorismo), labiopalatoschisi, tachicardia fetale, cardiopatie (difetti settali, ritorno venoso polmonare anomalo, atresia aortica, atresia mitralica, stenosi polmonare), dita delle mani flesse e sovrapposte. Meno frequentemente si osservano onfalocele, ernia ombelicale, golf ball, piedi torti, anomalie renali.
La biochimica permette di rilevare una riduzione nel siero materno della concentrazione di beta-hCG libera e di PAPP-A.

L'analisi citogenetica prenatale permette di rilevare un cromosoma 13 in sovrannumero o la traslocazione.
RISCHIO DI RICORRENZA
Il rischio di ricorrenza della trisomia 13 nelle famiglie con precedente feto affetto è di circa l'1%; nelle famiglie nelle quali la trisomia 13 si associa ad una traslocazione il rischio di ricorrenza è più elevato, specie se un genitore è portatore di una traslocazione bilanciata.
FENOTIPO
Neonato di basso peso con cranio trigonocefalico deficit di sviluppo delle bozze frontali), aree di aplasia cutanea al cuoio capelluto, labiopalatoschisi, microftalmia, ipotelorismo di grado variabile sino alla ciclopia (espressione di una sequenza oloprosencefalica), polidattilia postassiale, sindattilia, solco palmare unico, piede a gondola (tallone prominente e pianta del piede convessa), criptorchidismo nei maschi. Sono spesso presenti malformazioni cardiache e renali. I soggetti affetti in genere non superano l'anno di vita; quelli che sopravvivono oltre presentano un grave deficit di sviluppo.
Bibliografia
Demirel G, Oguz SS, Celik IH, et al., A trisomy 13 case with Robertsonian translocation presenting with atypical findings in Genet. Couns., vol. 21, nº 3, 2010, pp. 2937.
Hall AL, Drendel HM, Verbrugge JL, et al., Positive cell-free fetal DNA testing for trisomy 13 reveals confined placental mosaicism in Genet. Med., marzo 2013.
Janvier A, Farlow, Wilfond, The experience of families with children with trisomy 13 and 18 in social networks. in Pediatrics, vol. 130, nº 2, 23 luglio 2012, pp. 293298.
Patau K, Smith DW, Therman E, Inhorn SL, Wagner HP, Multiple congenital anomaly caused by an extra autosome in Lancet, vol. 1, nº 7128, 1960, pp. 7903
Peroos S, Forsythe E, Pugh JH, Arthur-Farraj P, Hodes D, Longevity and Patau syndrome: what determines survival? in BMJ Case Rep, vol. 2012, 2012.
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...