![]()
Approccio alla diagnosi ecografica delle Displasie Scheletriche
APPROCCIO ALLA DIAGNOSI ECOGRAFICA DELLE DISPLASIE SCHELETRICHE
Dr. P. Parisella
Le Displasie Scheletriche rappresentano un gruppo di anomalie genetiche legate alla mutazione di 364 geni comprendenti 436 condizioni cliniche raggruppate in 42 gruppi la cui classificazione(clicca qui) è pubblicata in allegato all'articolo di Bonafè L, Cormier-Daire V, Hall C. et al.: Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet Part A 167 (12):2869-2892.2015
Lo studio ecografico dello scheletro fetale può essere eseguito dalla 13a alla 30a settimana di gestazione.
Un corretto approccio alla diagnosi ecografica delle displasie scheletriche richiede due condizioni essenziali:
1) Esperienza dell'ecografista
2) Utilizzo di apparecchiature adeguate
Dal punto di vista diagnostico la variabilità fenotipica e le caratteristiche morfologiche non sempre consentono una diagnosi certa, ne tantomeno una diagnosi precoce (es. l'acondroplasia può essere spesso riconosciuta solo nel III trimestre). La diagnosi prenatale delle displasie scheletriche è più facile in presenza di una storia familiare positiva in quanto molte patologie sono ereditate con modalità autosomica recessiva. Nel sospetto di una displasia scheletrica è opportuno seguire un protocollo ecografico che prevede principalmente lo studio delle ossa lunghe, mani e piedi, torace, cranio, colonna vertebrale e l'eventuale associazione di anomalie di altri organi ed apparati.
La valutazione ecografica del feto con sospetta displasia scheletrica deve prevedere le seguenti tappe:
1)Valutazione presenza/assenza di uno o più arti o di uno o più segmenti ossei
2)Misura di tutte le ossa lunghe
a.Micromelia
b.Rizomelia
c.Mesomelia
d.Acromelia
3)Studio dei segmenti acromelici
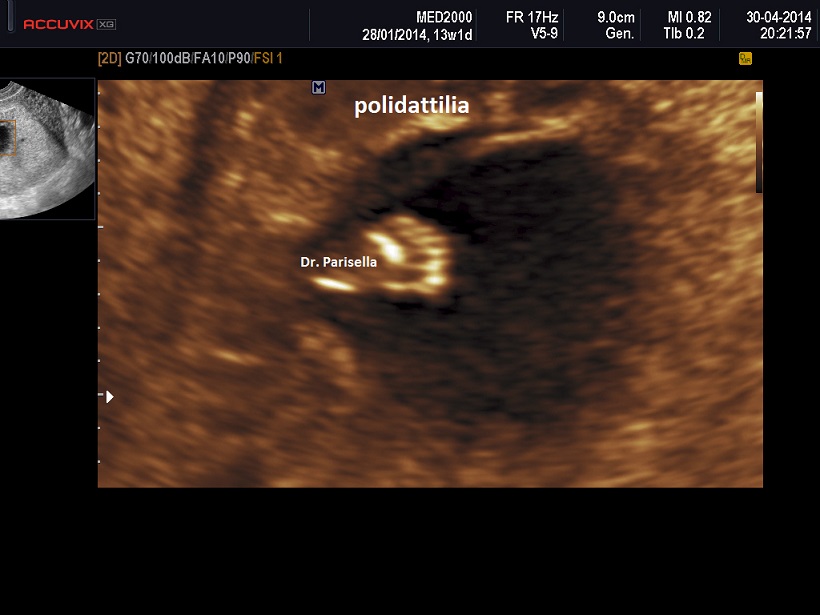
a.Polidattilia
b.Sindattilia
c.Deformità: clinodattilia, ectrodattilia, camptodattilia
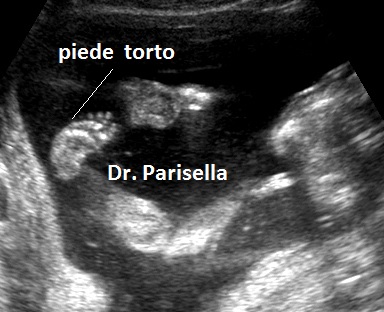
d.Piede torto
e.Mani torte
4)Aspetto dei segmenti ossei
a.Demineralizzazione
b.Incurvamento
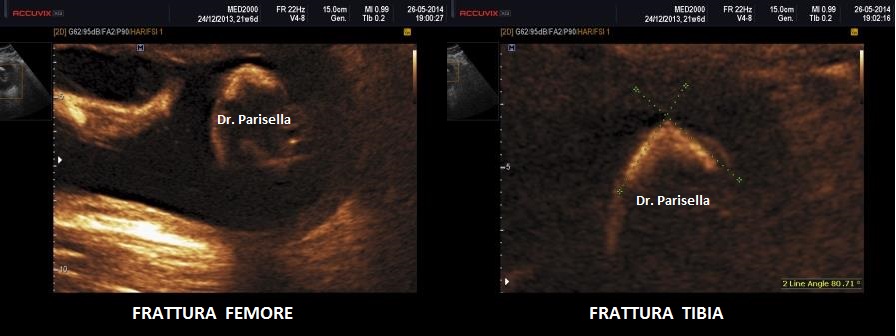
c.Fratture
d.Aspetto delle metafisi (slargamento)
5)Colonna Vertebrale
a.Demineralizzazione
b.Assenza/Ipoplasia sacro
c.Schisi
d.Platispondilia
e.Emivertebre
f. Malallineamento della colonna
6) Cranio/Faccia
a.Biometria
b.Macrocrania
c.Bozze Frontali (Frontal Bossing)
d.Radice del naso infossata
e.Schisi labiale e/o palato
f.Iper/ipo-telorismo
g.Cranio a trifoglio
h.Micrognazia
i.Craniosinostosi
l. NT I trimestre
7) Torace
a.Biometria (ipoplasia, torace stretto)
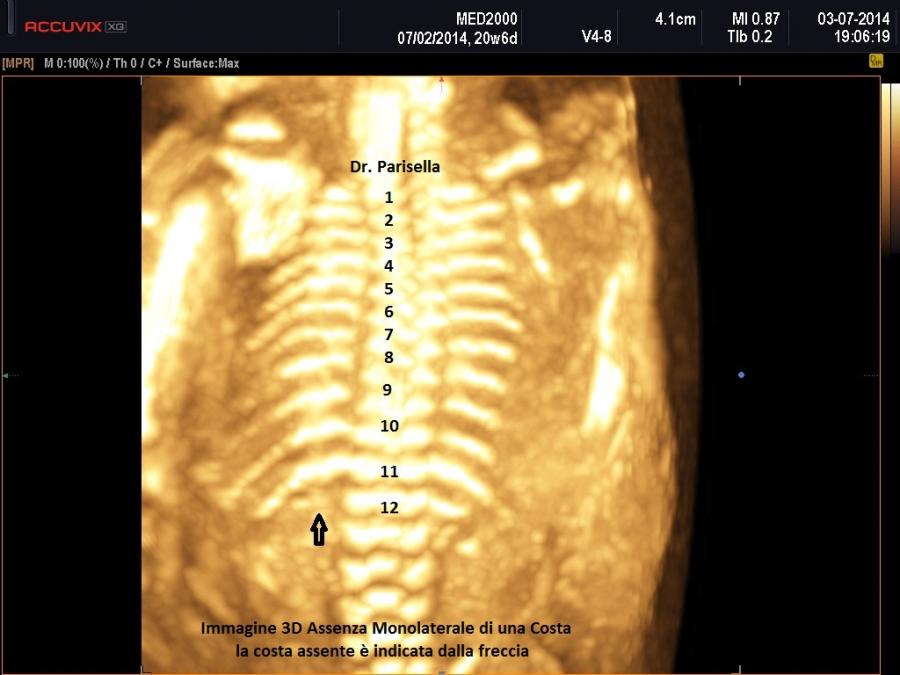
b.Coste: corte , ricurve, fratture
8) Studio di altri organi ed apparati
9) Studio accurato del cuore fetale (Ecocardiografia Fetale)
10) Studio dei movimenti fetali
11) Fattori prognostici
12) Consulenza genetica, esame del cariotipo, test molecolari se disponibili.
Per quanto riguarda il primo punto lo studio deve comprendere in modo dettagliato tutti e quattro gli arti e/o i loro segmenti: tratto rizomelico (femore-omero), tratto mesomelico ( tibia-perone ; radio-ulna), tratto acromelico (mano-piede) e la unilateralità o bilateralità.
Questi difetti sono in genere facilmente diagnosticabili; difficoltà possono aversi nello studio dell'arto posto distalmente o in presenza di condizioni come l'oligoidramnios e/o la gemellarità.
L'assenza completa di un arto è definita Amelia, di tutti e quattro Tetraamelia.
Si definisce Focomelia un difetto che interessa il tratto rizomelico e mesomelico con il tratto acromelico attaccato al tronco.
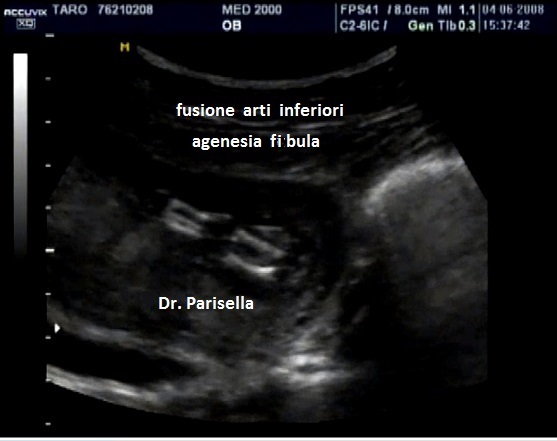
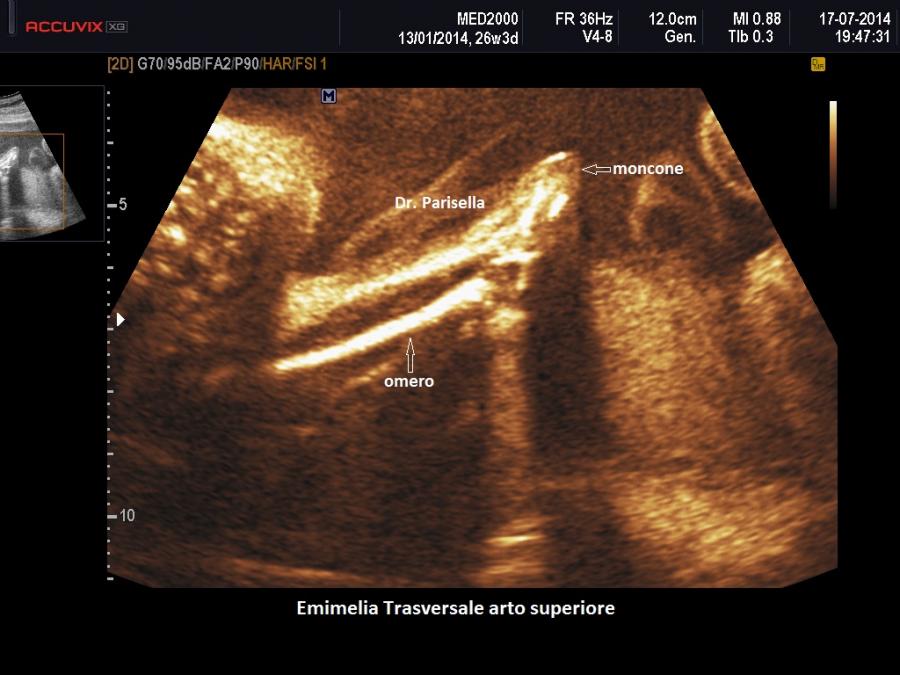
L'Emimelia è un difetto che interessa i segmenti mesomelico e acromelico. Può essere classificata in due tipi: 1) longitudinale quando a mancare è il raggio osseo mediale o laterale di un arto, cioè manca uno tra radio e ulna o uno tra tibia e perone; di solito radio e perone sono i più colpiti; nell'emimelia radiale si associa una mano torta; 2) trasversale quando manca completamente la parte distale di un arto: in questi casi in genere è presente un moncone tipo amputazione.
L'Acheiria (assenza della mano) e l'Apodia (assenza del piede) sono difetti che interessano il solo segmento acromelico.
La Sirenomelia, nella sua forma Monomelica, è caratterizzata dalla presenza di un solo arto inferiore localizzato in sede mediana.
Una volta accertata la presenza o assenza degli arti o di un segmento degli stessi si passa alla misurazione di tutte le ossa lunghe, previa conoscenza dell'esatta epoca gestazionale. Se ciò non fosse possibile si confronta il diametro biparietale o la circonferenza cranica con la lunghezza delle ossa lunghe; questo rapporto può essere accettato solo per quelle condizioni che non interferiscono con la crescita della testa. In genere si ritengono nella norma valori di lunghezza delle ossa lunghe compresi tra il 5° e il 95° percentile.Una eccezione è rappresentata dalla acondroplasiadove la misurazione degli arti si attesta intorno al 5° percentile e solo alla fine del II trimestre o allinizio del III si assiste ad un rallentamento della crescita.
A seconda dei segmenti ossei interessati le displasie vengono distinte in:
Displasia Micromelica: l'accorciamento interessa tutti i segmenti ossei;
Displasia Rizomelica: l'accorciamento interessa solo il femore e/o l'omero; spesso è associata a cromosomopatia.
Displasia Mesomelica: l'accorciamento interessa la tibia e/o il perone e/o il radio e/o l'ulna.
Displasia Acromelica: interessa la mano e/o il piede.
Ci soffermiamo ora sul femore. Un femore corto all'ecografia di screening del II trimestre può essere una normale caratteristica del feto, può essere la spia di un ritardo di crescita intrauterino del feto (IUGR), può essere un marker di aneuploidia. Un femore corto non isolato può essere il segno di vari quadri malformativi come le displasie scheletriche o essere uno dei segni di un'aneuploidia, principalmente trisomia 13 e trisomia 18.
La misurazione del femore ed il calcolo di alcuni rapporti possono essere di aiuto nello studio delle displasie scheletriche. Il calcolo del rapporto femore/piede non è molto utilizzato in quantotale rapporto può essere alterato maggiormente nelle displasie letali come l'Osteogenesi Imperfetta tipo II e la Displasia Tanatofora, rispetto a forme meno gravi evidenziabili più tardivamente come l'Acondroplasia. In caso di Acondroplasia può essere effettuata la misurazione dell'angolo femorale, anche se è una metodica non di comune esecuzione; recenti studi hanno riportato che nei feti con acondroplasia i processi patologici di ossificazione portano ad un aspetto caratteristico delle metafisi che tendono ad essere inclinate verso l'esterno determinando un aumento dell'angolo femorale.
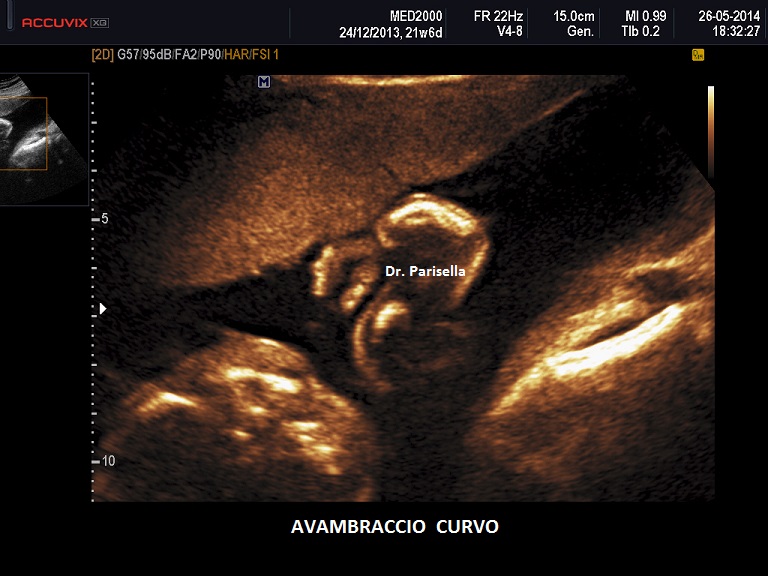
I quattro arti devono essere studiati anche dal punto di vista qualitativo, oltre che biometrico, valutando la presenza o l'assenza di mineralizzazione, curvature, fratture, slargamento delle metafisi.
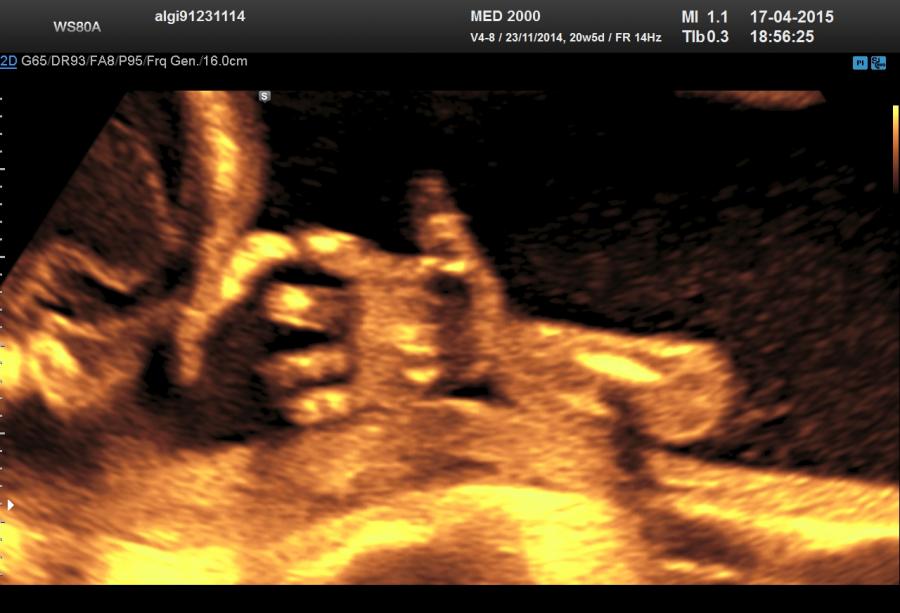
Le alterazioni a carico del tratto acromelico (di difficile o impossibile riconoscimento ecografico) consistono in polidattilia (preassiale, postassiale), sindattilia, clinodattilia, ectrodattilia, campodattilia.
Lo studio del tratto acromelico rappresenta un valido aiuto nella definizione delle varie displasie scheletriche per la frequente associazione tra tipo di anomalia del segmento acromelico e varie displasie. Tali patologie sono appresso elencate:
Polidatilia - viene distinta in:
Polidattilia preassiale se le dita sovrannumerarie sono localizzate al lato radiale per l'arto superiore e tibiale per l'arto inferiore (praticamente il dito sovrannumerario è contiguo al I dito).
Polidattilia postassiale se le dita sovrannumerarie sono localizzate al lato ulnare per l'arto superiore e fibulare per l'arto inferiore (praticamente il dito sovrannumerario è contiguo al V dito).
La Polidattilia è frequente nelle seguenti patologie: Associazione Water; Trisomia 21; Trisomia 13; Ipocondroplasia; S. di Jeune; S. Idroletale; Sindrome di McKusick-Kaufman (è autosomica recessiva con idrometrocolpo congenito, polidattilia, cardiopatia, utero bicorne e malrotazione intestinale); Sindrome di Bardet-Biedl (autosomica recessiva; va sospettata in caso di reni aumentati di volume, iperecogeni, polidattilia; Sindromi costa corta polidattilia tipo Majewski; Sindrome OFD I.
Polidattilia Preassiale si ritrova in: Disostosi Spondilocostale; Sindrome di Dubowitz.
Polidattilia Postassiale si ritrova in: Sindrome di Meckel-Gruber; Sindrome di Ellis-Wan-Creveld; Sindrome coste corte polidattilia tipo Saladino Noonan; Sindrome OFG II; Sindrome C.
Sindattilia - La sindattilia è causata dalla fusione delle ossa o dei tessuti molli delle dita adiacenti. E frequente nelle seguenti patologie: S. di Apert ; S. di Carpenter; S. di de Lange (secondo e terzo dito dei piedi); S. di Escobar; S. di Goltz; S. di Holt-Oram; S. di Jarcho-Levin; S. di Miller; Sindrome Oculo-dento-digitale (sindattilia di quarto e quinto dito delle mani, terzo e quarto dei piedi); Sindrome Oro-facio-digitale; S. di Pfeiffer; S. di Poland; S. di Roberts; Sindromedi Russel-Silver (secondo e terzo dito dei piedi); S. di Saethre-Chotzen; Sindrome di Smith-Lemli-Opitz (secondo e terzo dito dei piedi); Triploidia (terzo e quarto dito delle mani); Trisomia parziale 10q;Criptoftalmia.
Clinodattilia è caratterizzata dalla deviazione laterale di un dito sul piano palmare. E frequente nelle seguenti patologie: S. di Aarskog; S. di de Lange; S di Escobar; S. di Langer-Giedion; S. di Mohr; S. di Roberts; S. di Ruvalcaba; S. di Saethre-Chotzen; S. di Seckel; Sinostosi multiple; S. di Down; Trisomia 4p; Trisomia 9p; Trisomia 20p; XXXX; XXXXX; XXXXY; XXY; 13q-
Ectrodattilia è caratterizzata dall'assenza di un singolo dito sino ad arrivare, nelle forme più gravi, all'assenza di tutte le dita tranne il quinto. Comprende un gruppo di anomalie delle mani e dei piedi come nella Split Hand/Foot dovuta all'assenza del terzo dito con schisi che si approfonda verso la parte prossimale della mano o del piede e sindattilia delle dita ai lati della schisi e conseguente tipico aspetto a chela di aragosta.
Campodattilia è caratterizzata dall'atteggiameno in flessione delle articolazioni interfalangee prossimali; a volte si associa un ispessimento connettivale della superficie volare. Si riscontra nella trisomia 13; trisomia 18; 1q+; 4p+; trisomia 8; trisomia 9; S. di Pena-Shokeir; Disostosi Spondilocostale; etc.
Bisogna valutare anche le alterazioni dell'asse tra segmento mesomelico e quello acromelico; tra queste rara è l'alterazione dell'asse tra avambraccio e mano (mano torta) mentre è frequente l'alterazione dell'asse tra gamba e piede ( Piede Torto ) che può essere espressione di varie patologie quali: spina bifida, idrocefalia, cromosomopatia (es. trisomia 18,.....), oligoidramnios, etc
..
Bisogna poi valutare anche il grado di mineralizzazione delle ossa, che riguarda non solo le ossa lunghe ma anche altri distretti come il cranio e il rachide.
La ipomineralizzazione la ritroviamo: 1) Osteogenesi Imperfetta Tipo II: sono interessate tutte le ossa;
2) Ipofosfatasia: sono interessate tutte le ossa;
3) Acondrogenesi: sono interessati calvario e rachide.
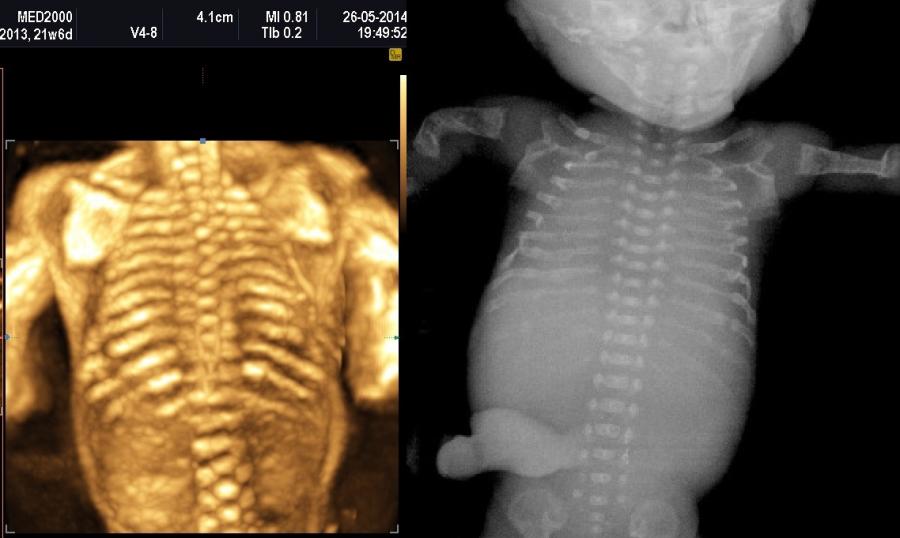
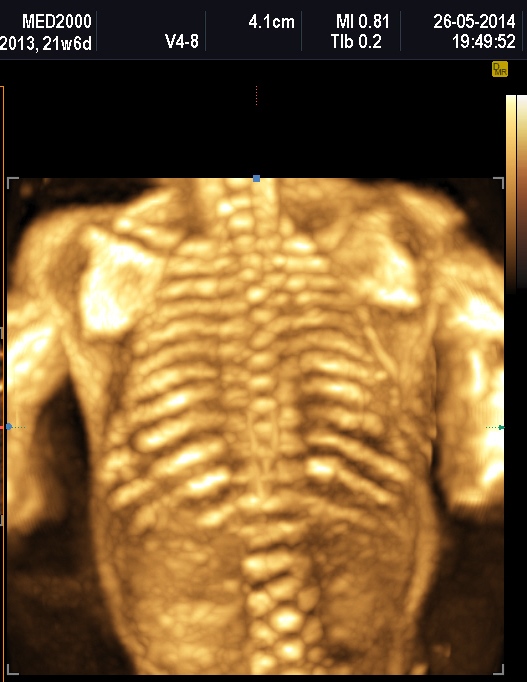
Le displasie caratterizzate da ipomineralizzazione, specie se spiccata, possono presentare fratture dei segmenti ossei interessati.
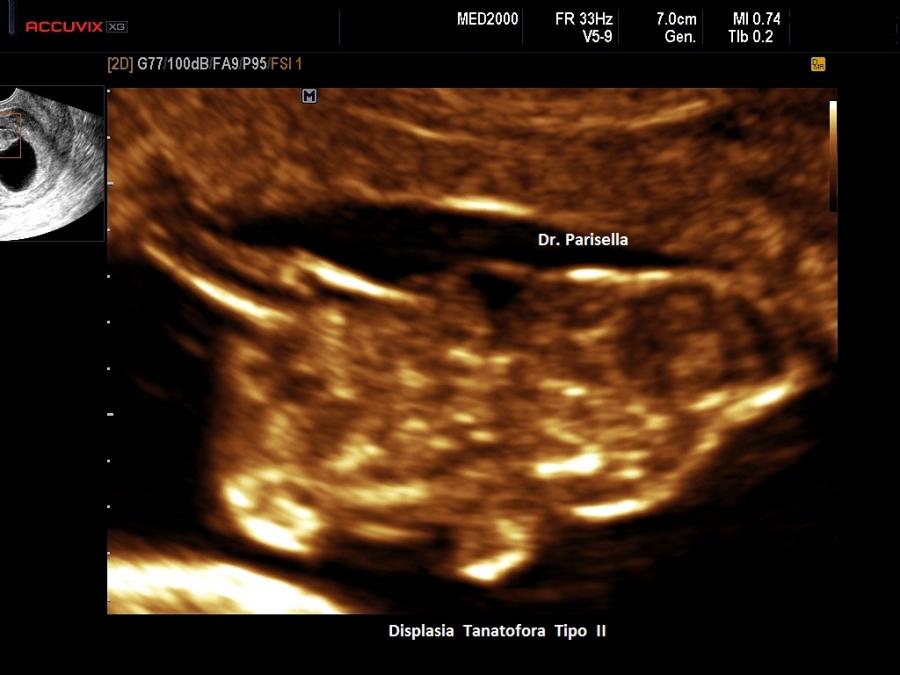
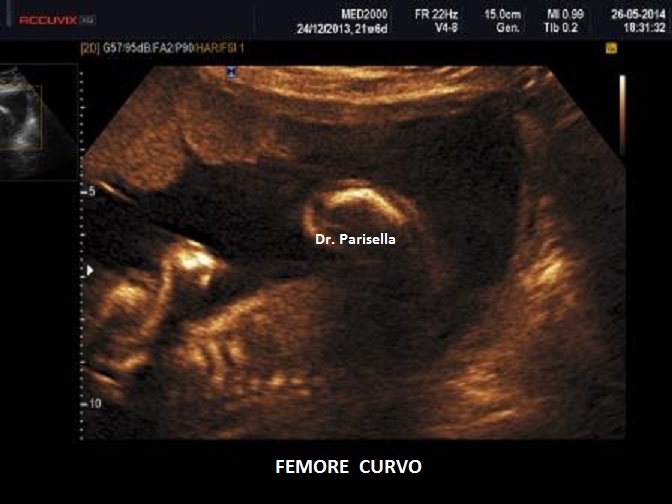
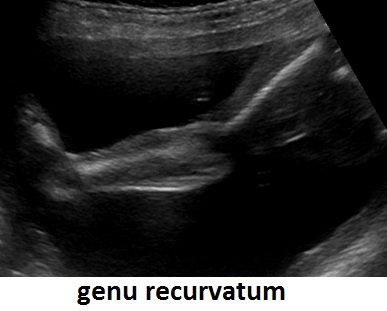
Un altro dato da valutare è il grado di curvatura ossea; le due principali condizioni che possono presentare in epoca prenatale un incurvamento delle ossa lunghe sono l'osteogenesi imperfetta e la displasia tanatofora tipo I (femori a cornetta di telefono).
Per quanto riguarda il rachide la Platispondilia (comunque non facilmente evidenziabile ecograficamente) è presente nella Displasia Tanatofora e nella Displasia Metatropica. Nell'Acondrogenesi tipo I si ha scarsa ossificazione dei corpi vertebrali e costantemente non si visualizza il sacro.
La misurazione dei diametri del torace è di notevole importanza nella valutazione di un feto con displasia ossea. La scansione di riferimento per la valutazione della biometria del torace fetale e dell'anatomia dei campi polmonari è la scansione assiale delle 4-camere cardiache; per la misurazione della lunghezza toracica ci si avvale della scansione sagittale-paramediana-destra.
La misura della circonferenza toracica con un valore < 5° percentile è un indice di ipoplasia polmonare, causa di distress respiratorio nel neonato e indice di letalità. La biometria toracica e la morfologia toracica possono orientarci verso una particolare forma di displasia: se il torace è eccessivamente piccolo potrebbe trattarsi di una Displasia Tanatofora; se il torace è lungo e stretto potrebbe trattarsi di una Sindrome di Jeune; se le coste sono molto corte potrebbe trattarsi di una Sindrome coste corte con o senza polidattilia; se ci sono fratture costali potrebbe trattarsi di una Osteogenesi Imperfetta tipo II; se le coste sono fuse tra loro potrebbe trattarsi di una Displasia Spondilocostale.
Vari Autori hanno proposto diversi parametri per valutare l'ipoplasia polmonare:
- rapporto circonferenza toracica/circonferenza addominale < 5° percentile
- rapporto area cardiaca/area toracica < 5° percentile
- rapporto torace/lunghezza del tronco < 0,32
- rapporto area polmonare destra/area toracica < 0,11
- diametro polmonare destro < 5° percentile
Anche la biometria cranica assume notevole importanza nell'inquadramento diagnostico delle displasie scheletriche.
Si intende per Macrocefalia/Macrocrania un aumento della circonferenza cranica al disopra del 97° percentile; si intende per Microcefalia una diminuzione della circonferenza cranica al disotto del 3° percentile.
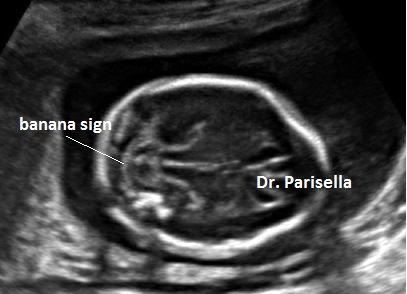
Macrocefalia/Macrocrania è presente nella Acondroplasia, nella Displasia Tanatofora ( il tipo II è caratterizzato da cranio a trifoglio ), nell'Acondrogenesi. L'idrocefalia può essere una complicazione dell'Acondroplasia, dell'Osteogenesi Imperfetta e della Displasia Metatropica. Un cranio di consistenza molle (ossa wormiane) è sintomo costante di Osteogenesi Imperfetta anche se può essere presente in altre condizioni quali l'ipofosfatasia, la displasia cleido-cranica, etc. Nell'Ipofosfatasia vi può essere una aumentata ecogenicità della falce cerebrale.
Notevole importanza assume lo studio della faccia: profilo fetale, bozza frontale, naso, mandibola, orbite.
Inoltre bisogna valutare l'eventuale interessamento di altri organi ed apparati: Polidramnios e Idrope fetale si associano frequentemente alle displasie scheletriche soprattutto con l'Acondrogenesi e l'Osteogenesi Imperfetta. Cardiopatie Congenite si trovano spesso nella Displasia Condro-Ectodermica e nelle Sindromi costa corta polidattilia. Il rene policistico è frequente nella Displasia Toracica Asfissiante. Un discorso a parte deve essere fatto sulla prognosi delle displasie scheletriche dove assume notevole importanza clinica la distinzione tra forme letali e non letali. Una accurata ecografia prenatale può essere in grado di predire la prognosi nella maggioranza dei casi. Uno dei rapporti biometrici utilizzati è il rapporto lunghezza del femore/circonferenza addominale: se è < 0,16 può indicare una forma letale. Allo stesso modo l'ipoplasia polmonare che segue ad un torace piccolo e/o stretto suggerisce una prognosi infausta; non sempre però una ipoplasia toracica è espressione di una forma letale. Entrano in gioco anche altri fattori come la presenza di idrope fetale, grave polidramnios, malattie di altri organi ed apparati associate alla displasia, principalmente le anomalie cardiovascolari, cerebrali e renali che contribuiscono ad aggravare il quadro clinico. Le forme non letali sono un gruppo più eterogeneo che interessano principalmente gli arti e le estremità e meno frequentemente possono essere diagnosticate in epoca prenatale precoce; l'esempio classico è l'acondroplasia dove la biometria degli arti può essere normale fino a tutto il II trimestre e il difetto di crescita delle ossa lunghe si rende manifesto solo nel III trimestre o alla nascita.
Di fronte al sospetto di una displasia scheletrica bisogna essere molto cauti poichè spesso una diagnosi precisa può essere formulata solo in epoca postnatale. Può essere utile allo scopo tener presenti alcune regole:


Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...