![]()
Distrofia Miotonica Distrofia miotonica tipo 1 (DM1) o Malattia di Steinert OMIM 160900 Distrofia Miotonica tipo 2 (DM2) o PROMM (PROximal Myotonic Myopaty) OMIM 602668
Distrofia miotonica
Distrofia miotonica tipo 1 (DM1) o Malattia di Steinert OMIM 160900
Distrofia Miotonica tipo 2 (DM2) o PROMM (PROximal Myotonic Myopaty) OMIM 602668
redatto da P.Parisella
- distrofia miotonica tipo 1 (DM1) o Malattia di Steinert
- distrofia miotonica tipo 2 (DM2) o PROMM (PROximal Myotonic Myopaty)
- occhi: cataratta
- cuore: aritmie, cardiomiopatie
- sistema endocrino: ipotiroidismo, diabete
- apparato riproduttivo: ridotta fertilità
- sistema nervoso centrale: ritardo mentale, disturbi del comportamento
IL CASO
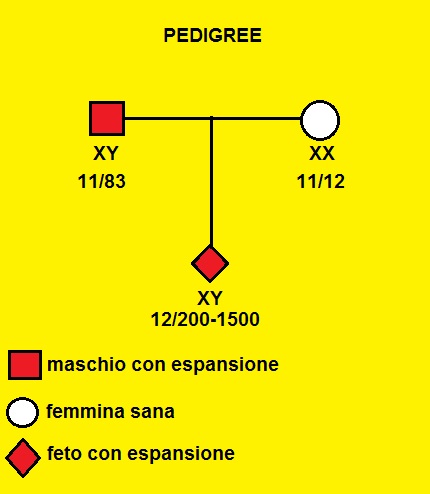
Paziente con anamnesi positiva per casi di Distrofia Miotonica in consanguinei del padre del nascituro.
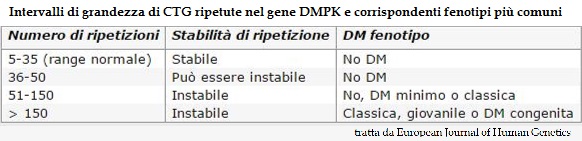
L'analisi genetica dei prodotti di amplificazione ha dato i seguenti risultati:
- madre XX: presenza di due prodotti di amplificazione contenenti circa 11 e 12 ripetizioni CTG rispettivamente;
- padre XY: presenza di due prodotti di amplificazione contenenti circa 11 e 83 ripetizioni CTG rispettivamente;
- il DNA estratto dai villi coriali mostra la presenza oltre a un prodotto di amplificazione normale contenente 12 ripetizioni CTG (di origine materna), di un allele espanso, verosimilmente di origine paterna, contenente da 200 a 1500 ripetizioni CTG .
Conclusioni: l'esame eseguito sul DNA estratto dai villi coriali prelevato dalla paziente XX ha mostrato la presenza in eterozigosi nel nascituro di un allele espanso contenente da 200 a 1500 ripetizioni CTG nella regione 3' non codificante del gene DMPK: tale genotipo è compatibile con la Distrofia Miotonica di Steinert.
L'esame ecografico eseguito alla 17a settimana di gestazione ha mostrato un feto con normale attività cardiaca e normale attività motoria; la morfologia fetale, compatibilmente con l'epoca gestazionale, appariva nella norma; normale e compatibile con l'epoca di amenorrea l'accrescimento fetale; normale per aspetto e quantità il liquido amniotico; normale l'inserzione e l'ecostruttura della placenta.
Antonio Cao, Malattie genetiche. Molecole e geni. Diagnosi,
prevenzione e terapia, Padova, Piccin [2004], febbraio 2004.
Arsenault,
M.E., Prevost, C., Lescault, A., et al. Clinical characteristics of myotonic
dystrophy type 1 patients with small CTG expansions. Neurology 66: 1248-1250,
2006.
Bundey, S.
Clinical evidence for heterogeneity in myotonic dystrophy. J. Med.
Genet. 19: 341-348, 1982.
Censori, B., Provinciali, L., Danni, M., et al. Brain involvement in myotonic
dystrophy: MRI features and their relationship to clinical and cognitive
conditions. Acta Neurol. Scand. 90: 211-217, 1994.
Donahue, L.
A., Mangla, R., Westesson, P.-L. Neuroimaging in myotonic dystrophy type 1.
Neurology 73: 1931 only, 2009.
Ekström AB,
Hakenäs-Plate L, Samuelsson L, Tulinius M, Wentz E.., Autism spectrum
conditions in myotonic dystrophy type 1: a study on 57 individuals with
congenital and childhood forms. in Am J Med Genet B Neuropsychiatr Genet.,
147B, 2008, pp. 918-926.
Groh WJ,
Groh MR, Saha C, et al.: Electrocardiographic abnormalities and sudden death in
myotonic dystrophy type 1. in N Engl J Med., vol. 358, giugno 2008, pp.
2688-2697.
Kamsteeg
EJ, Kress W, Catalli C, et al. Best
practice guidelines and recommendations on the molecular diagnosis of myotonic
dystrophy types 1 and 2. European Journal of Human Genetics (2012) 20, 12031208
Steinberg H, Wagner A., Hans Steinert: 100 years of myotonic dystrophy in Cardiol Prat., vol. 79, agosto 2008, pp. 961-70.
Tramonte,
J. J., Burns, T. M. Myotonic dystrophy. Arch. Neurol. 62: 1316-1319, 2005.
Zeesman,
S., Carson, N., Whelan, D. T. Paternal transmission of the congenital form of
myotonic dystrophy type 1: a new case and review of the literature. Am. J. Med.
Genet. 107: 222-226, 2002.
Zuhlke, C.,
Roeder, E., Purmann, S., et al. Homozygous myotonic dystrophy: clinical
findings in two patients and review of the literature. Am. J. Med. Genet. 143A:
2058-2061, 2007.
Aggiornamenti
- Patologie Genetiche dello Scheletro
Sono elencate tutte le 436 Displasie Scheletriche con la... - Consigli per l'utilizzo del software diagnosi in Diagnosi...
Consigli per l'utilizzo del software diagnosi in Diagnosi... - TERMINI DI USO DEL PORTALE WEB med2000eco e Software...
TERMINI DI USO DEL PORTALE WEB med2000eco e Software...